Überraschender Rückzug: Warum das Krebsmedikament Amivantamab vom Markt verschwand
Pharmafirma Janssen-Cilag nahm 2022 das zugelassene Krebsmedikament Amivantamab vom deutschen Markt. Das entscheidende Gremium sah keinen Zusatznutzen. Ob Amivantamab für Patient:innen ein lebenswichtiges Medikament ist, darüber streiten sich die Fachleute.
Überraschend nahm die Pharmafirma Janssen-Cilag im vergangenen August das bereits zugelassene Krebsmedikament Amivantamab vom deutschen Markt. Wenige Wochen zuvor hatte der Gemeinsame Bundesausschuss – das höchste Gremium der Selbstverwaltung im deutschen Gesundheitswesen – entschieden: Es liegen nicht genug Daten vor, um einen Zusatznutzen in der Krebstherapie zu belegen. Das Beispiel zeigt, dass auch nach der Zulassung die Validierung neuer Medikamente in Deutschland nicht abgeschlossen ist.
Amivantamab wirkt nur bei wenigen Menschen
Amivantamab ist ein biotechnologisch erzeugter Antikörper, der als Medikament zur Behandlung von Erwachsenen mit einer bestimmten Form von Lungenkrebs, dem nicht-kleinzelligen Lungenkrebs (NSCLC), im fortgeschrittenen Stadium zugelassen ist. Im Mai 2021 erteilte die US-amerikanische Arzneimittelbehörde FDA die Zulassung, die europäische Zulassungsbehörde EMA folgte am 9. Dezember desselben Jahres. Vermarktet wird Amivantamab unter dem Handelsnamen Rybrevant von der US-amerikanischen Johnson-&-Johnson-Pharmasparte Janssen Biotech. In Deutschland führt die Geschäfte die Janssen-Cilag GmbH mit Sitz in Neuss.
Amivantamab ist allerdings nicht für alle NSCLC geeignet, sondern für einen genetischen Sonderfall. Wenn nämlich eines der beiden Zielproteine des therapeutischen Antikörpers an einer bestimmten Stelle mutiert ist. Fachleute sprechen von einer Exon-20-Insertionsmutation im epidermalen Wachstumsfaktorrezeptor (EGFR).
Dieser genetische Subtyp des NSCLC betrifft nur etwa ein bis zwei Prozent der Lungenkarzinom-Erkrankten, also rund 500 Menschen pro Jahr. Und nur ein weiterer Bruchteil kommt für eine Therapie mit Amivantamab in Frage, da der Antikörper bislang nur zur Zweitlinientherapie zugelassen ist. Das bedeutet, dass zuvor eingesetzte Chemotherapeutika nicht ausreichend gewirkt haben.
Die Spezifität des Antikörpers ist Fluch und Segen zugleich. Denn für Betroffene ist Amivantamab ein sehr genaues und dadurch nebenwirkungsärmeres Werkzeug beim Kampf gegen den Krebs. Dass es aber nur wenige Betroffene gibt, ist gleichermaßen ein Schwachpunkt. Dazu später mehr.
AMNOG: Nach der Zulassung erfolgt eine weitere Prüfung
Bevor ein Medikament in der EU zugelassen werden kann, prüft die Europäische Arzneimittel-Agentur (EMA), ob es wirksam und sicher ist. Nur dann darf es verkauft werden und wird in Deutschland von den Krankenkassen erstattet. Diese Hürde nahm Amivantamab auf Basis einer Phase-1-Studie, mit der Janssen zeigen konnte, dass der Antikörper das Leben der Proband:innen verlängert.
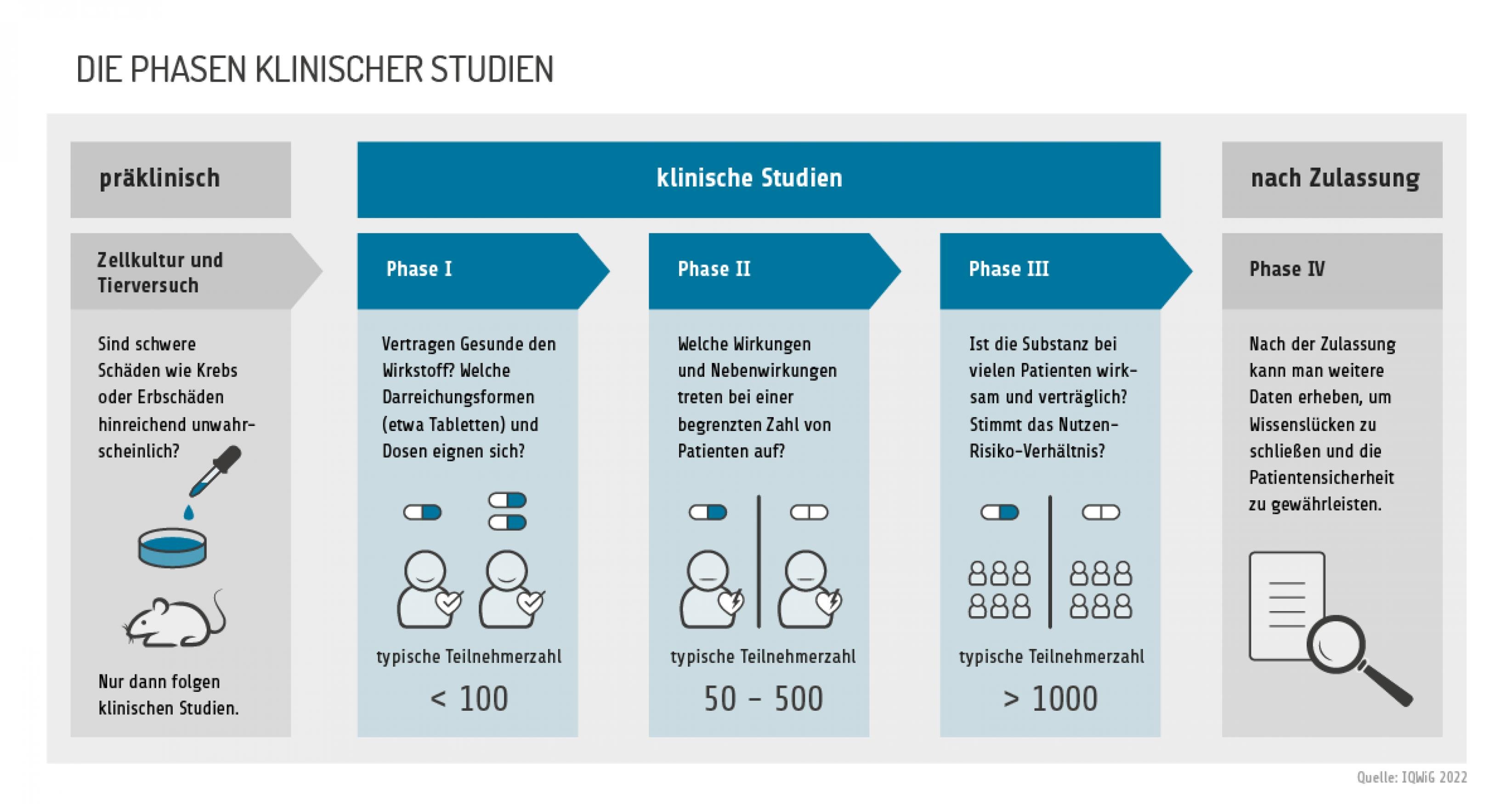
Seit 2011 gilt in Deutschland aber das Arzneimittelmarktneuordnungsgesetz, kurz: AMNOG. Dessen Ziel ist es unter anderem, die seit vielen Jahren stark steigenden Arzneimittelausgaben der gesetzlichen Krankenkassen zu deckeln. Dafür bewerten Gremien, ob ein neues Arzneimittel einen höheren Nutzen hat als bereits vorhandene Therapieoptionen. Im Falle von Amivantamab ist das die Chemotherapie.
Das letzte Wort hat dabei der Gemeinsame Bundesausschuss (G-BA). Er beauftragt das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) mit der Nutzenbewertung.
Die Zusatznutzen-Bewertung ist nicht nur eine zusätzliche bürokratische Hürde, es geht um viel Geld. Laut IQWiG sparten die gesetzlichen Krankenkassen im Jahr 2019 durch das AMNOG-Verfahren mehr als drei Milliarden Euro ein. In den ersten zwölf Monaten nach Markteintritt kann der Hersteller nämlich den Preis für sein Medikament frei festlegen. Im Falle von Amivantamab und Janssen-Cilag bedeutete das: 135.000 Euro, pro Jahr und Patient.
Stellen die Gremien allerdings keinen Zusatznutzen fest, zahlen Krankenkassen nur noch den Preis, den ein Standard-Präparat kosten würde. Damit muss sich die Pharmafirma zufriedengeben – oder sie zieht das Präparat zurück.
Kein Zusatznutzen für Amivantamab
Mit der Zulassung durch die EMA musste sich also auch Amivantamab im AMNOG-Verfahren beweisen. Dieses begann am 15. Januar 2022. Rund sechs Monate später – am 7. Juli 2022 – entschied der G-BA: „Zusatznutzen nicht belegt.“
Es fehlten schlichtweg Daten. „Um einen aussagekräftigen Vergleich zu erreichen, sollten Patientinnen und Patienten den beiden Therapieoptionen nach dem Zufallsprinzip zugewiesen werden und die Ergebnisse der Behandlung verglichen werden“, schreibt Volker Vervölgyi, Bereichsleiter im IQWiG-Ressort Arzneimittelbewertung, auf Anfrage.
Eine solche randomisierte, vergleichende Studie hatte der Amivantamab-Hersteller zu diesem Zeitpunkt aber noch nicht durchgeführt. In Phase 2 der klinischen Prüfung setzte Janssen Amivantamab allein oder in Kombination mit anderen Therapien ein. Eine Vergleichs-Proband:innen-Gruppe fehlte, nämlich Menschen, die zwar Chemotherapeutika, aber kein Amivantamab erhalten hatten.
Zusatznutzen nicht belegt.
G-BA zum Krebsantikörper Amivantamab
Um dennoch vergleichen zu können, zog Janssen Daten von zwei Lungenkrebs-Registern hinzu. Eine davon verwaltet molekulare und klinische Daten von mehr als 20.000 Patient:innen und ist eine der größten Lungenkrebs-Datenbanken weltweit.
Bei dem Vergleich kam laut Janssen heraus, dass mit Amivantamab behandelte Lungenkrebs-erkrankte Menschen fast doppelt so lange überlebten wie Menschen, die eine klassische Therapie erhalten hatten, und zwar rund 23 statt 12 Monate.
Nicht ausreichend, urteilten IQWiG und G-BA: „Der Bewertung lag […] ein nicht randomisierter Vergleich zugrunde“, schreibt Vervölgyi. Dadurch seien die Daten von vornherein mit einer hohen Unsicherheit behaftet. Daran änderte auch der Vergleich mit den Registerdaten nichts.
Die Entscheidung des G-BA heißt also nicht, dass es keinen Zusatznutzen für Amivantamab gibt. Die Gremien entschieden, dass nicht ausreichend Daten vorhanden sind, um einen möglichen Zusatznutzen überhaupt zu beurteilen.
Fachgesellschaften kritisieren die G-BA-Entscheidung
Das kann Frank Griesinger nicht nachvollziehen: „Von medizinischer Seite waren wir schon davon überzeugt, dass die Daten ausreichen müssten, um zumindest einen nicht quantifizierbaren Zusatznutzen zu generieren“, sagt er. Der Direktor der Klinik für Hämatologie und Onkologie der Oldenburger Universitätsmedizin hat an zahlreichen Leitlinien zur Behandlung von Lungenkrebs mitgearbeitet. (Hinweis: Griesinger erhielt Honorare von Janssen und war an wissenschaftlichen Untersuchungen beteiligt, die von Janssen finanziert wurden.)
Griesinger ist außerdem Vorsitzender des Arbeitskreises Lungenkarzinom der Deutschen Gesellschaft für Hämatologie und Medizinische Onkologie (DGHO). Die DGHO kommentierte gemeinsam mit anderen Fachgesellschaften die Entscheidung des G-BA in einer Stellungnahme.
Wir sehen keine Möglichkeit, Rybrevant weiterhin in Deutschland zur Verfügung zu stellen.
Janssen-Cilag
Ein Argument, welches sowohl Fachgesellschaften als auch Hersteller anführen: Randomisierte, klinische Studien lassen sich mit so kleinen Patientengruppen nur schwer durchführen. Der G-BA hingegen schreibt auf Anfrage, dass auch für Arzneimittel mit kleinen Patientenkollektiven durchaus qualitativ anspruchsvolle Studien möglich seienund verweistauf das Blutkrebs-Medikament Blinatumomab. Am 20. Januar 2022 stufte der G-BA „den Zusatznutzen des Wirkstoffs Blinatumomab für ein neues, sehr seltenes Anwendungsgebiet als erheblich“ ein.
Anderthalb Monate nach der Entscheidung des G-BA nahm Janssen-Cilag Amivantamab am 24. August 2022 mit sofortiger Wirkung vom deutschen Markt.
Auf Nachfrage schreibt der Pharma-Hersteller: „Wir sind uns der Verantwortung für die Versorgung der Patient:innen bewusst und bedauern sehr, dass diese nun Rücksprache mit ihren behandelnden Ärzt:innen halten müssen, um über ihre weiteren Behandlungsoptionen zu beraten.“
Am Medikamenten-Rückzug ändert das aber erst einmal nichts. „Wir haben alle Möglichkeiten geprüft, Rybrevant als Innovation weiterhin in Deutschland zur Verfügung zu stellen. Auf Basis des Nutzenbeschlusses des G-BA sehen wir hierzu jedoch keine Möglichkeit“, schreibt Janssen-Cilag.
Medikamenten-Rückzug: Wie geht es weiter?
Die Leidtragenden seien die an Krebs erkrankten Menschen, sagt Frank Griesinger. Sowohl die Bewertung durch die Gremien als auch das Zurückziehen des Medikaments durch das pharmazeutische Unternehmen nennt er nicht nachvollziehbare Entscheidungen. „Die Folge ist, dass Patienten nicht mehr sicher mit dem Medikament versorgt werden“, sagt er. Hinzu kommt eine große Verunsicherung bei Ärzt:innen und Patient:innen, besonders solchen, die bereits eine der mehrere Monate dauernden Therapien begonnen haben.
Sicherlich könnten die behandelnden Ärzte und Onkologinnen Amivantamab im Ausland ordern, denn auf den Rest der EU hat Janssens Deutschland-Rückzug keinen Einfluss. Aber das bringe hohen administrativen Aufwand mit sich, ist Griesinger überzeugt. Es sei unklar, wer die Kosten für die Therapie übernimmt; es müssten umfangreiche Kostenübernahme-Anträge gestellt und von medizinischen Diensten geprüft werden. All das seien nicht nur riesige Hürden, sondern verzögere den gesamten therapeutischen Prozess.
Unklar ist aber, für wie viele Menschen in Deutschland die Marktrücknahme tatsächlich bedeutsam ist. Der Hersteller selbst schätzt im Rahmen des AMNOG-Verfahrens, dass bis zu 130 Patient:innen jährlich von der EGFR Exon-20-Insertionsmutation bei NSCLC betroffen sind. Bis zu 29 von ihnen sollen laut Janssen nach Versagen einer platinbasierten Therapie für eine weitere Therapie infrage kommen. Das IQWiG rechnet zwar mit deutlich höheren Zahlen. Dennoch ist die Zahl der Menschen, für die eine Behandlung mit Amivantamab infrage kommt, gering.
Hinzu kommt, dass auch eine Therapie mit dem Antikörper mit Nebenwirkungen einhergeht. Der EMA-Bewertungsbericht etwa führt Hautausschlag, Ödeme, Übelkeit und Erbrechen sowie Erkrankungen der Lunge und infusionsbedingte Reaktionen auf.
Transparenzhinweis: Die Autorin hat mögliche Interessenkonflikte des zitierten Experten ergänzt sowie im letzten Absatz die Experteneinschätzung gekürzt und um kontrastierende Informationen ergänzt. (31. März 2023)
Dieser Beitrag basiert auf einem Artikel, der imMärz 2023 im Laborjournalerschienen ist.